| Taxon names | Citations | Turn highlighting On/Off |






(C) 2012 R. Henrik Nilsson. This is an open access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
For reference, use of the paginated PDF or printed version of this article is recommended.
Molecular data form an important research tool in most branches of mycology. A non-trivial proportion of the public fungal DNA sequences are, however, compromised in terms of quality and reliability, contributing noise and bias to sequence-borne inferences such as phylogenetic analysis, diversity assessment, and barcoding. In this paper we discuss various aspects and pitfalls of sequence quality assessment. Based on our observations, we provide a set of guidelines to assist in manual quality management of newly generated, near-full-length (Sanger-derived) fungal ITS sequences and to some extent also sequences of shorter read lengths, other genes or markers, and groups of organisms. The guidelines are intentionally non-technical and do not require substantial bioinformatics skills or significant computational power. Despite their simple nature, we feel they would have caught the vast majority of the severely compromised ITS sequences in the public corpus. Our guidelines are nevertheless not infallible, and common sense and intuition remain important elements in the pursuit of compromised sequence data. The guidelines focus on basic sequence authenticity and reliability of the newly generated sequences, and the user may want to consider additional resources and steps to accomplish the best possible quality control. A discussion on the technical resources for further sequence quality management is therefore provided in the supplementary material.
ITS, sequence reliability, sequence quality control, fungi, databases, barcoding
The inconspicuous and largely subterranean or endophytic nature of much of fungal life presents a challenge to mycology. Many fungal lineages do not seem to produce tangible fruiting bodies, and for those that do, the factors promoting - and acting against - fruiting body formation are only partly understood. As a result, most sampling sites and habitats host a much greater fungal diversity than the above-ground view offered by fruiting bodies would lead the observer to believe (
For all their advantages, molecular data do not solve all open research questions in mycology, and examples of where the misuse and misinterpretation of molecular data hampered mycological progress are easy to point out (
The most popular genetic marker for mycological research questions at and below the genus level is the nuclear ribosomal internal transcribed spacer (ITS) region, a ca. 450–650 base pair (bp.) region consisting of the two variable spacers ITS1 and ITS2 and the intercalary, highly conserved 5.8S gene (
Overview of the five guidelines.
| Target of guideline | Way of getting there |
|---|---|
| 1. Establish that the sequences come from the intended gene or marker | Do a multiple alignment of the sequences and verify that they all feature some suitable, conserved sub-region (here the 5.8S gene) |
| 2. Establish that all sequences are given in the correct (5’ to 3’) orientation | Examine the alignment for any sequences that do not align at all to the others; re-orient these; re-run the alignment step; and examine them again |
| 3. Establish that there are no (bad cases of) chimeras in the dataset | Run the sequences through BLAST in INSD/UNITE and verify that the best match comprises more or less the full length of the query sequences |
| 4. Establish that there are no other major technical errors in the sequences | Examine the BLAST results carefully, particularly the graphical overview and the pairwise alignment, for anomalies |
| 5. Establish that any taxonomic annotations given to the sequences make sense | Examine the BLAST hit list to see that the species names produced make sense |
We would like to stress that the guidelines described here focus on basic sequence authenticity and reliability; they are certainly no panacea for sequence quality management. Their purpose is to assist in pruning severely compromised entries from newly generated, nearly full-length (typically, but not exclusively, Sanger-derived) fungal ITS datasets before those sequences are put to scientific use. The target audience comprises researchers who have just started to use molecular tools (e.g., students) as well as those who otherwise would have taken little action in the direction of quality management. For the user wishing to apply the most advanced and technical quality control solutions to a new dataset right from the start, we provide an account of the bioinformatics of ITS sequence quality control in Appendix. One is nevertheless mistaken to believe that sequence reliability is a matter of bioinformatics only; taxonomic knowledge and common sense are just as important, if much more difficult to algorithmize. What follows is an attempt at a joint treatment of these three aspects.
A word on the query and reference datasetsThe sequences in INSD and UNITE are often used as reference datasets to which newly generated (“query”) sequences are compared in pursuit of taxonomic and ecological annotation. Neither INSD nor UNITE seek to store full ITS sequence datasets generated by next-generation sequencing (NGS) technologies such as 454 pyrosequencing (
Much of the following will apply also to genes and markers other than the ITS region – particularly the neighbouring ribosomal small subunit (SSU) and large subunit (LSU) genes - and it will certainly apply to the ITS region in groups of organisms other than fungi. Nevertheless, for the sake of example, the user is assumed to have a newly generated fungal ITS dataset (with chromatograms), ideally of near-full-length sequences or at least sequences covering approximately the same part of the ITS region. A proportion of the sequences is assumed to be annotated to various hierarchical classification levels, such as “Uncultured chytridiomycete”, “Penicillium sp.”, and “Amanita muscaria”. To avoid overly simplified examples, we will furthermore assume that the data offer some degree of taxonomic complexity and span several fungal phyla and multiple orders. If the dataset is small - say fewer than 50 sequences - the user should probably consider each sequence individually. For datasets up to a few hundred sequences, the user could use a clustering tool such as the BLASTclust implementation at http://toolkit.tuebingen.mpg.de/blastclust to reduce the dataset to one representative sequence per “species” or operational taxonomic unit (OTU;
Upwards of five hundred public sequences are, or have previously been, annotated as ITS sequences when they in fact have been shown to represent other genes or markers or are noise (seemingly random nucleotide letters) throughout. The reasons could be many and range from primer matches to unexpected parts of the genome at hand to the mixing up of test tubes, files, or individual sequences. These sequences contribute significant noise to any data-mining effort targeting the fungal ITS sequence corpus by, e.g., inflating diversity estimates. For molecular identification of fungi, these sequences pose something of an indirect problem, since they are very unlikely to show up in ITS-based BLAST searches (
An expedient way to ensure that all query sequences represent the ITS region is to compute a multiple sequence alignment in any of a number of on-line multiple alignment services, notably MAFFT (http://mafft.cbrc.jp/alignment/server/ ;
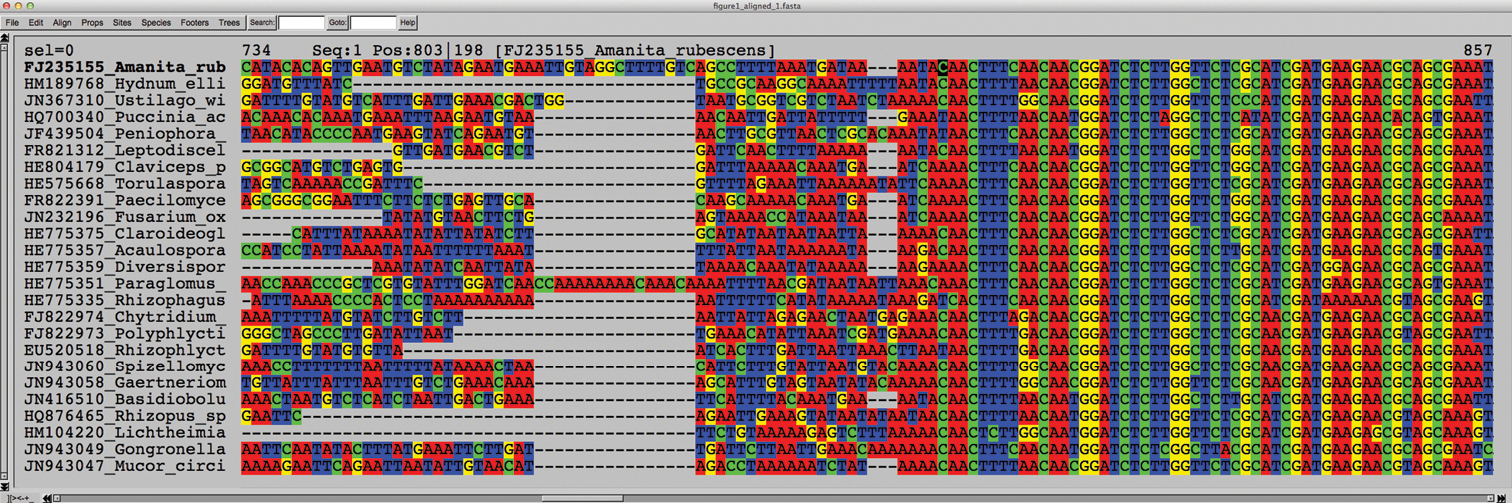
An ITS alignment featuring five random species each of the fungal phyla Ascomycota, Basidiomycota, Glomeromycota, Chytridiomycota, and Zygomycota s.l. The left half of the screen represents the ITS1 and the right half the 5.8S. Whereas the ITS1 alignment appears more or less chaotic, the 5.8S stands out as a very conserved element throughout these five phyla. The 5.8S starts at position 803 (indicated by the black cursor in the uppermost sequence). Seaview (
Sequences that do not produce any noteworthy similarity to the 5.8S region of the alignment are likely to belong to one of four categories: 1) they may be partial ITS sequences, containing nothing, or very little, of the 5.8S; 2) they may represent genes or markers other than the ITS (comprising, for example, the 3’ SSU intron); 3) they may be of very low read quality or even feature random sequence data altogether; and 4) they may be reverse complementary. The case of reverse complementary sequences is handled separately below (Guideline 2); for the other three - and for the few fungi with truly divergent 5.8S/ITS region sequences, such as Cantharellus and Tulasnella (
As an alternative to the alignment-based approach, the user may choose to subject the query sequences - individually or, more likely, in batches - to BLAST searches in INSD. Whether or not a sequence is an ITS sequence can usually be inferred from the annotation of the top five matches alone. As a rule of thumb, a high-quality fungal ITS sequence that features the full 5.8S gene will always produce at least 100 ITS-related BLAST (blastn) matches of a bitscore of about 200 or greater (if only to the 5.8S itself) in INSD under default settings. A sequence that, in contrast, produces just a handful of matches most certainly requires further scrutiny and is, in our experience, very unlikely to qualify as a high-quality ITS sequence in the end.
Guideline 2. A single alignment step can assess the orientation of the query sequencesWhile it perhaps would seem natural to assume that all newly generated sequences come in the correct (5’ to 3’) orientation, this is in practice not always the case. A study by
It would seem likely that most reverse complementary sequences are produced during the contig assembly, a semi-to-fully-automated step where the sequence data produced by each primer employed are brought together to form the full sequence – a contig (cf.
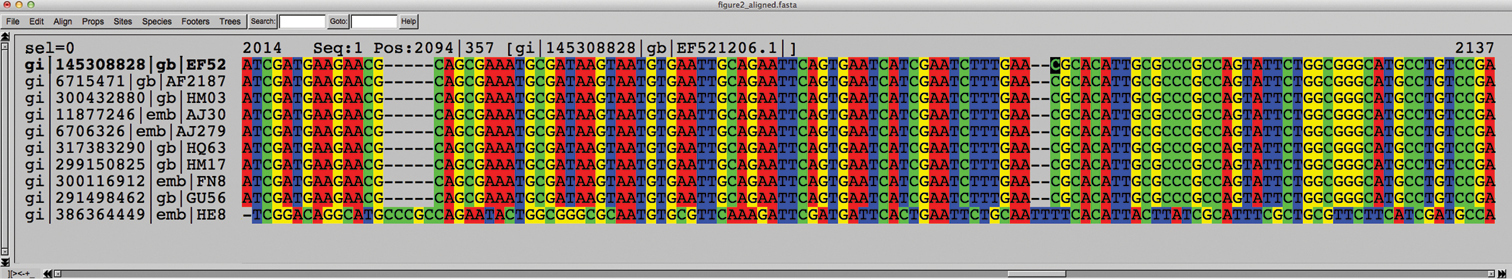
A reverse complementary sequence (bottom) aligned to its nine best BLAST matches, all of which were nearly identical to the query sequence based on BLAST scores, and all of which were given in the correct orientation by their respective authors.
An alternative, and perhaps less advisable, approach to reverse complementary control involves BLAST in INSD. By default, BLAST offers native support for reverse complementary queries (as well as reference sequences) and makes very little noise if a reverse complementary sequence is found. In fact, the user has to scroll down several pages of BLAST output - to the actual alignment produced by BLAST - to get an idea of whether a query sequence is reverse complementary or not. Here, the item “Strand=Plus/Plus” indicates that both the query and the reference sequence are in the same read direction. If the five to ten best matches are all “Strand=Plus/Plus” (and particularly if they come from two or more different studies), the user can be reasonably certain that the query sequence is given in the correct orientation. Similarly, several consecutive “Strand=Plus/Minus“ suggest that the query sequence is reverse complementary (Figure 3). Problematically, but logically, a reverse complementary sequence in INSD will produce a “Strand=Plus/Plus” BLAST result to a reverse complementary query, with the second match hopefully showing “Strand=Plus/Minus“. In other words, based on the BLAST output alone it is not always easy to conclude which sequence is reverse complementary and which is given in the regular orientation. Indeed, the hypothetical existence of large batches of reverse complementary INSD sequences for some particular species would interfere with the above observations, suggesting that the best way to approach reverse complementary control is by looking at the actual sequence data in a multiple alignment. A special case of reverse complementary sequences - the reverse complementary chimera - is treated under Guideline 4 below.
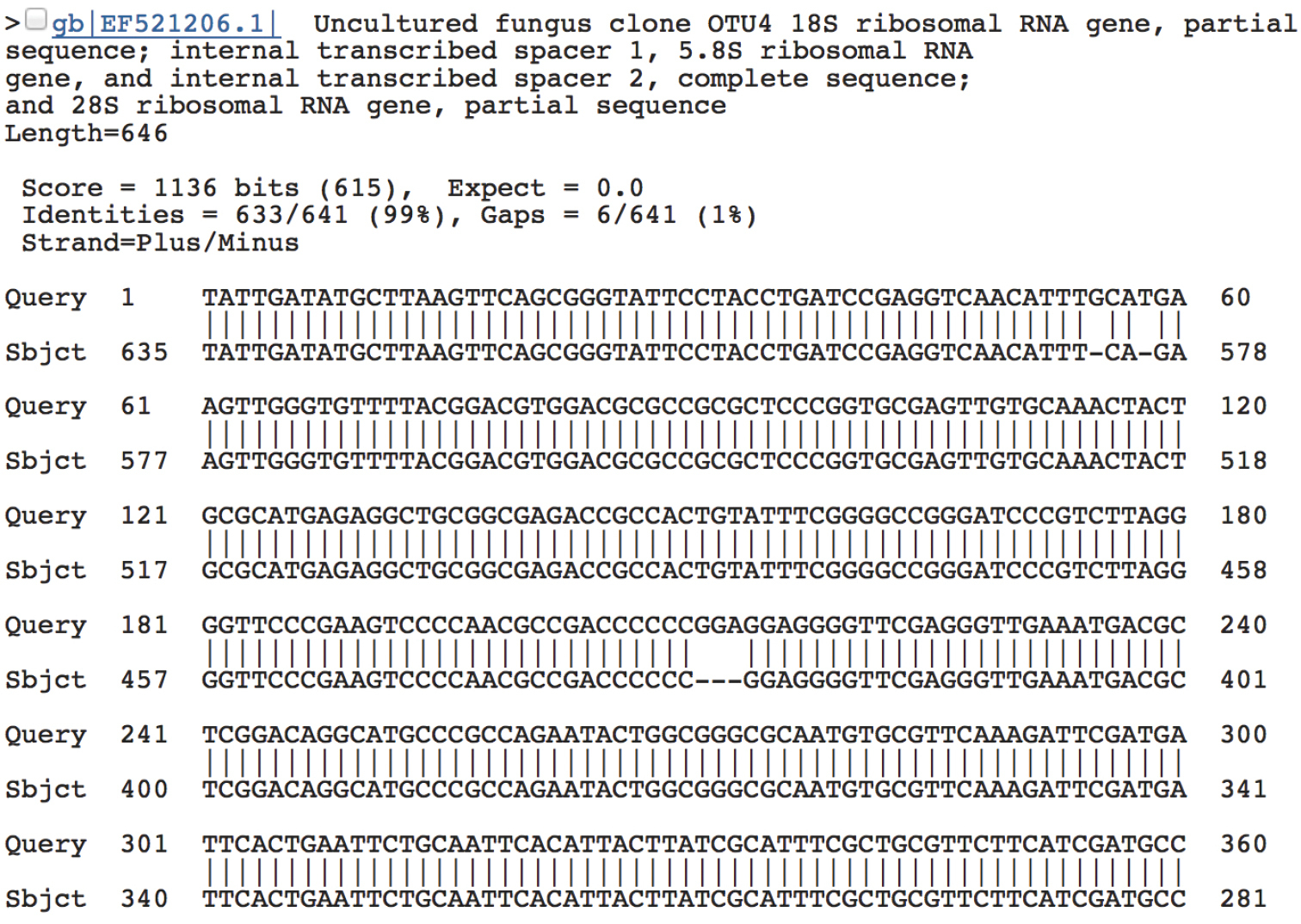
“Strand=Plus/Minus” indicates that the query and reference sequence come in opposing read directions. Another hint comes from the observation that the alignment starts at the first base (1) in the query sequence and progresses upwards to base 60 in the first alignment line; however, for the reference sequence, the alignment starts at base 635 and progresses downwards to base 578.
The traditional view of a PCR chimera is an artificial sequence resulting from the joining of two (or occasionally more) sequence fragments that do not originate from the same species (see Guideline 4 for a wider definition). In a typical fungal ITS chimera, either the ITS1 comes from one species and 5.8S plus ITS2 come from another, or ITS1 plus 5.8S come from one species and ITS2 from another (Figure 4). In other words, the chimeric breakpoint often seems to be located in the first – and more conserved - part of the 5.8S. These traditional chimeras can unintentionally be produced in the PCR step when the DNA of two or more species are present and when the gene or marker in question features a highly conserved segment (here the 5.8S; cf.

A multiple alignment where the topmost sequence is chimeric and the remaining sequences represent its best BLAST matches. The alignment is fine in ITS1 and 5.8S (a; the 5.8S starts at position 479), but the alignment in ITS2 (b; position 637 and on) falls far short of scientific rigour. Alignments like these bespeak chimeric unions.
UNITE has a record of about 1, 000 chimeric fungal ITS sequences in the public corpus, corresponding to 0.4% of the number of such sequences. The real number of chimeras is probably significantly higher, since chimeras between closely related species are much more difficult to find than chimeras between distantly related ones. The vast majority of the 1, 000 known chimeras are of the “distantly related” type; the chimera in Figure 4 is such an example. Cloning of PCR amplicons is a component in many studies in which chimeras were subsequently reported, suggesting that studies employing cloning should be particularly vigilant against chimeric unions. Fortunately, finding at least bad cases of chimeras in newly generated datasets is fairly straightforward. The solution draws from the observation that chimeric sequences tend to be unique in datasets of small to moderate sizes, i.e., that any given illegitimate union of sequence fragments happened only once in the study. This somewhat rough approximation means that the user can cluster the query dataset at approximately the species level (97-98% similarity, 90% sequence coverage; see above) and then focus on the singletons (or all small-sized OTUs) only. By subjecting the singleton sequences to BLAST searches and keeping an eye on the graphical summary of the BLAST hits provided by NCBI-BLAST (http://blast.ncbi.nlm.nih.gov/ ), the user will be able to identify sequences in need of further scrutiny. Figure 5a shows a BLAST run where a query sequence was well matched across its full length by the topmost hits. Figure 5b, in contrast, shows a chimeric sequence where the 5.8S and ITS2 were well matched by the topmost hit, whereas the ITS1 could not be aligned at all to it. This corresponds to the case where the ITS1 comes from a distantly related species with respect to the remainder of the sequence. All cases where ITS1+5.8S - or 5.8S+ITS2 - produce nearly perfect matches, whereas ITS2 or ITS1, respectively, produces an unexpectedly poor match, call for closer scrutiny.
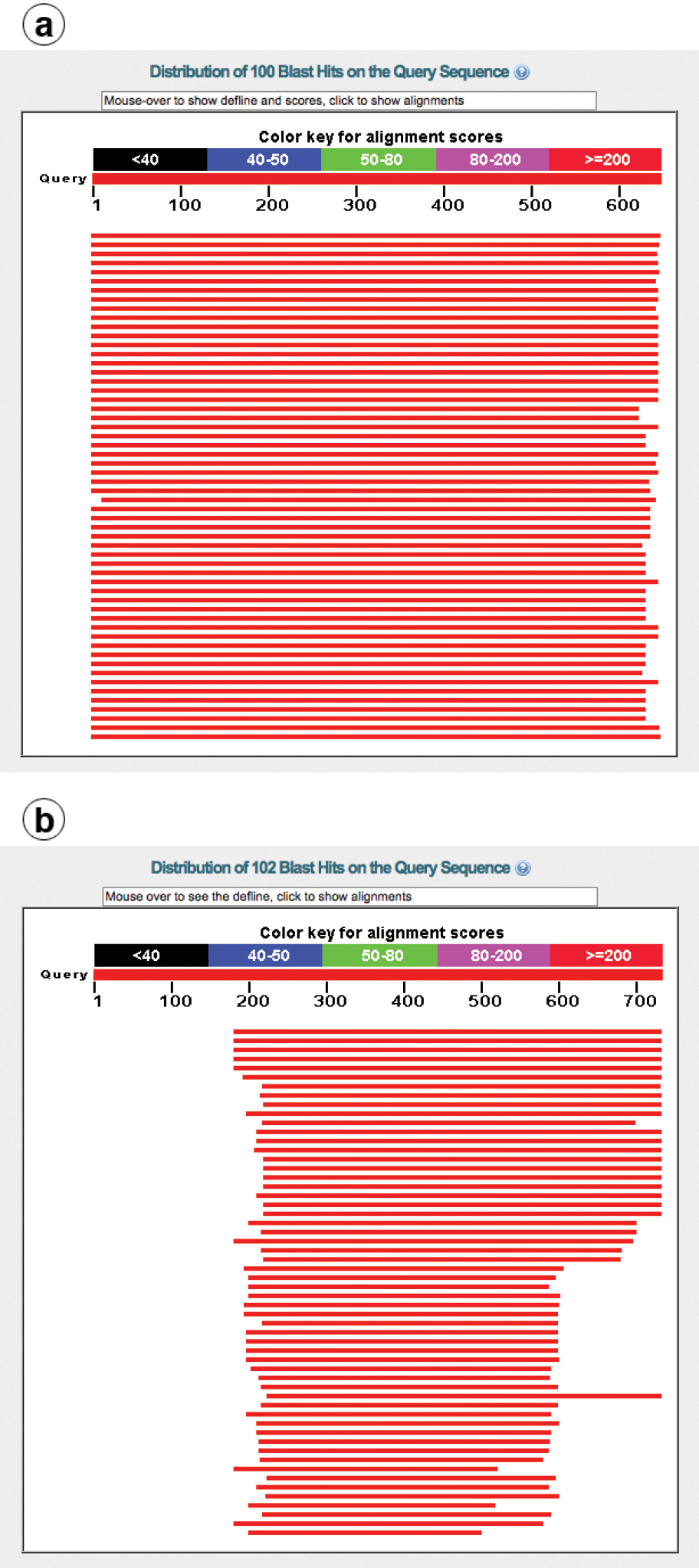
a Graphical overview of the BLAST results of a regular sequence b BLAST results of a chimeric sequence where the ITS1 comes from another species, such that the ITS1 is not involved in the alignment featuring the 5.8S+ITS2 (hence the lack of a match for the first ca. 180 bp.). Obviously, a severely compromised sequence that is already in INSD will always find a perfect match through BLAST in INSD: itself. In that case, the presence of a 100% similar reference sequence cannot be used as a testimony to the authenticity of the query sequence.
In the case of Figure 5b, it is the ITS1 that does not harmonize with the remainder of the sequence. Doing a BLAST search based on ITS1 alone shows that it is a polypore (100% similarity); the 5.8S+ITS2 BLAST, in contrast, shows that those parts belong to an agaric (100% similarity). By doing separate BLAST searches like this, the user will come fairly close to practical proof that the sequence in question is chimeric. Such sequences should be pruned from the query dataset, and they should similarly not be submitted to the sequence databases. However, the user should keep in mind that legitimate query sequences - particularly long ones - can also produce BLAST results similar to that in Figure 5b for the reason that the most similar reference sequences were much shorter due to, e.g., primer choice. The BLAST alignment indicates at what base in the query and the reference sequence the alignment starts. For example, if the alignment start is “1” in the reference sequence but “350” in the query sequence, then the seemingly odd BLAST results simply reflect the absence of reference data. Introns such as the one at the 3’ end of the SSU may produce similar results. However, also in these situations, subjecting the non-matching part of the query sequence to a BLAST search is likely to reveal the nature of the problem.
Problematically, not all cases of chimera detection will be as straightforward as the example in Figure 5b, and the user will sometimes face difficult decisions. After all, ITS sequence data are available for a mere 1% of the hypothesized 1.5 million extant species of fungi (
BLAST also has the capacity to indicate several other classes of compromised entries. Figure 6 shows an assembly chimera, which is the product of incorrect assembly of two or more sequence fragments (primer reads) into a single contig. The dotted vertical line in the reference sequences indicates a break in the alignment between these and the query sequence. The user will have to scroll down to the BLAST alignment to learn of the exact nature of the break. Often one finds that such sequences were assembled with the ITS1 and the ITS2 in the wrong order. The resulting BLAST alignment will be divided into sections, and the user might find that, e.g., base 285 to 614 in the query sequence are matched by bases 1 to 330 in the reference sequence. Bases 1-284 in the query are, however, best matched by bases 331-614 in the reference sequence; although it may not always be straightforward to see exactly what the problem is, the non-contiguous nature of these alignment segments at least makes it easy to see that there indeed seems to be a problem to begin with. If all alignment sections are in the Strand=Plus/Plus orientation, and the next few reference sequences similarly produced such sectioned alignments with respect to the query, then the user can be certain that the query sequence is an assembly chimera. It is easy to see that assembly chimeras may follow as a result of minimal overlap between the fragments under assembly and the subsequent failure of the contig software – under the settings applied - to pick the correct ends for merger. If there is no overlap at all between the fragments - such that there should have been additional sequence data between two fragments that are now joined - the corresponding BLAST results will look something like Figure 6. Such bridged sequences may also be produced inadvertently in, e.g., the phylogenetic analysis package PAUP (
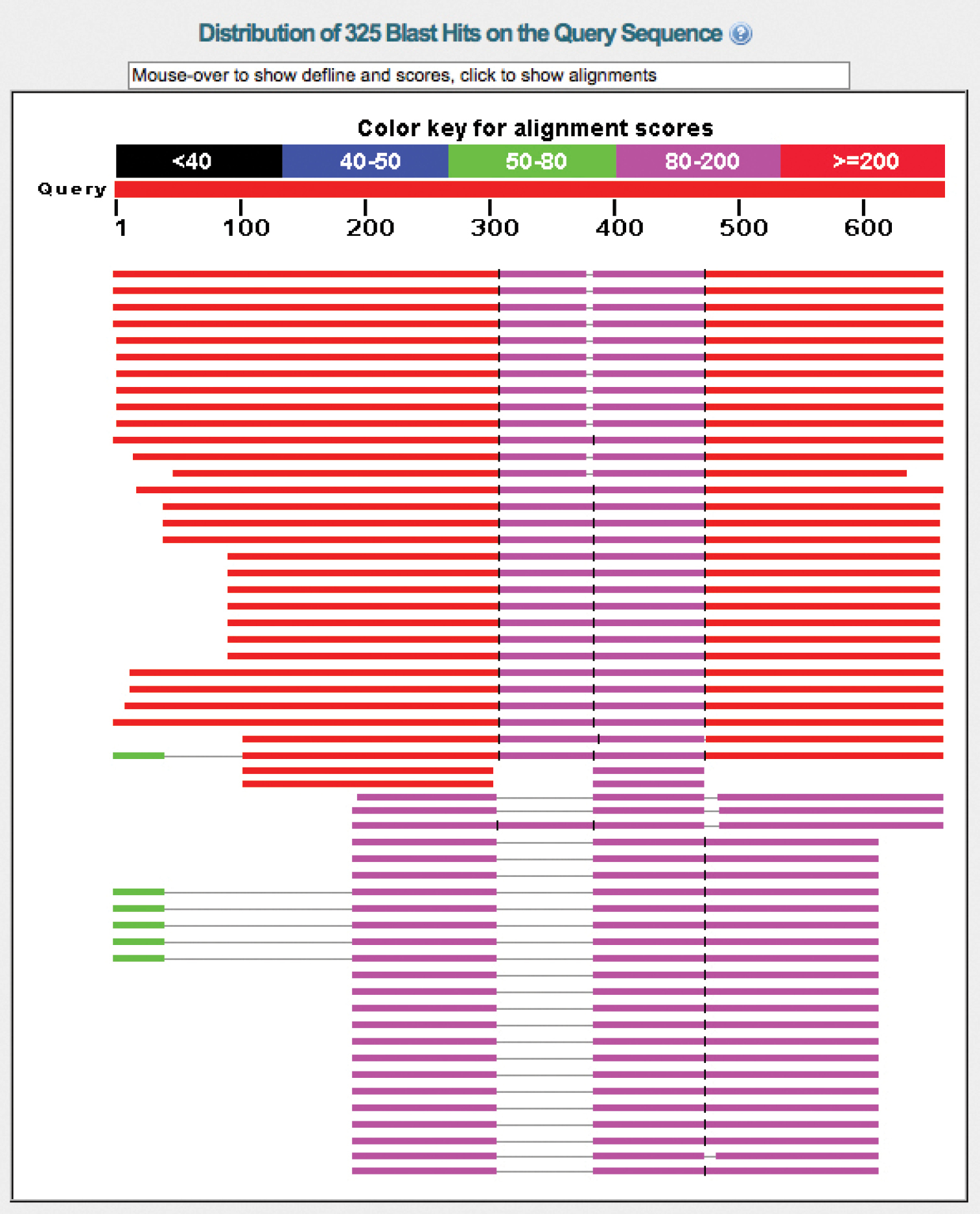
An assembly chimera. The black dashed lines indicate breaks in the BLAST alignment and should always be taken to mean that manual examination is needed.
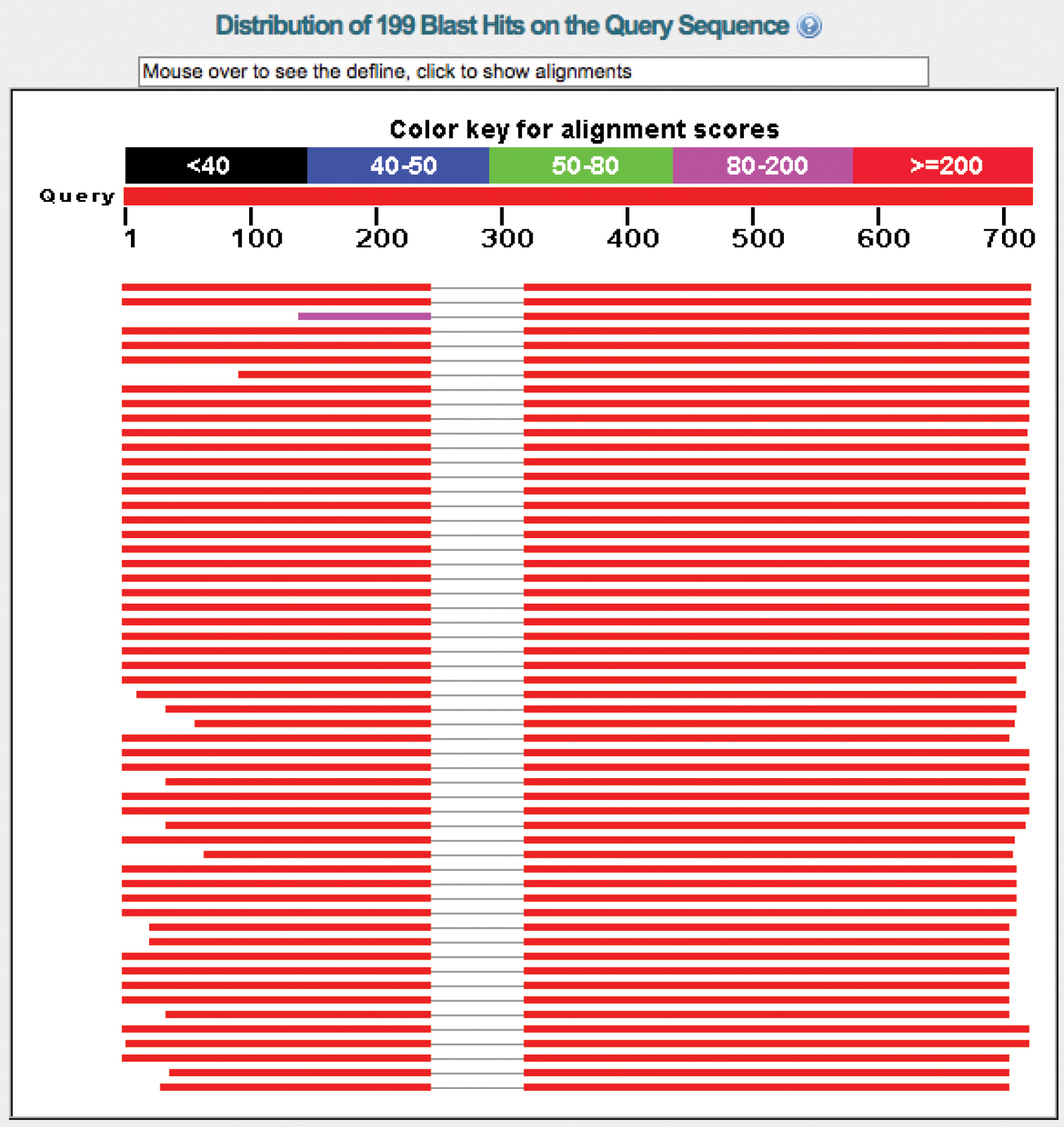
An assembly chimera. An extraneous sequence segment was assembled into a position where it should not have been, such as in the middle of the 5.8S. The white area in the reference sequences indicates the absence of sequence data for this particular part of the query sequence. Manual examination is always needed in cases like this.
The distal (5’ and 3’) ends of newly generated sequences are typically of lower read quality than the interior parts of the sequence. It is the job of the contig assembly software to highlight poorly read bases clearly enough that the user can address them before the final sequence is produced from the contig. Untrimmed sequences tend to look like the one in Figure 8 when run through BLAST; note that the match does not include the first ca. 20, and the last ca. 30, base-pairs. Unless all of the reference sequences are in fact shorter than the query sequence, the user should probably recheck the chromatograms in the distal parts of the sequence - and consider trimming regions of poor quality - at this stage. Many public ITS sequences, in turn, are poorly trimmed, sometimes leaving the process of telling whether it is the query or the reference sequence that features the low-quality bases all but intractable. This speaks to the importance of always taking the sequence assembly step seriously and of paying special attention to any region where the chromatograms appear substandard. Other newly generated sequences are of reduced read quality throughout. One obvious sign is that they may feature IUPAC DNA ambiguity symbols (e.g., N and S;

Untrimmed sequences tend to look like this when run through BLAST. Note how the first ca. 20 bp., and the last ca. 30 bp., of the query sequence (represented by the red bar with scale marks every 100 bp.) do not align to any of the BLAST hits. The use of different but closely situated primers may give a similar pattern, however, pointing at the need to also look at the BLAST alignments for start and end positions of the reference sequences.
About half of the 250, 000 public, full-length fungal ITS sequences are annotated to the level of species (
We take the position that all sequences in a newly generated dataset should be verified for taxonomic affiliation, even if they are annotated only to kingdom level (e.g., “Uncultured fungus”). The process of verifying a hypothesized taxonomic annotation - or at least ruling out the possibility that the annotation is way off - is usually trivial and amounts to a simple BLAST run. A sequence annotated as Penicillium is expected to hit other Penicillium sequences (usually in a chaotic list of anamorphic and teleomorphic names, species complexes, and numerous environmental sequences; a visit to Index Fungorum (http://www.indexfungorum.org/ ) or MycoBank (http://www.mycobank.org/ ) may be needed to establish the relations of the names obtained). A quick check of some degree of consistency among the top ten matches is normally enough to confirm the basic authenticity of the taxonomic affiliation, particularly if the top ten matches stem from two or more different studies. The INSD keyword “BARCODE” (specified in the description of the entry) indicates that a sequence complies with a number of quality criteria (http://barcoding.si.edu/pdf/dwg_data_standards-final.pdf ) and so should be weighted as a more reliable reference sequence. However, looking at BLAST hit lists is often more difficult than one might think. The following five basic principles may be good to keep in mind. a) BLAST is sensitive to the length and level of sequence conservation of the query and reference sequences, and the user is advised to prune any large parts of the SSU and LSU from the ITS sequences before doing BLAST searches (cf.
It is typically simple to establish basic authenticity of the taxonomic annotations for a set of query sequences. The process described above will often take the user to the genus level or even the species level in some cases, at which stage one can rule out severe misannotation. Going all the way to actually verifying the species-level annotation is a trickier objective, and one that will not always be possible based on BLAST and the public sequence databases alone. A phylogenetic analysis of the query sequence and the 20-30 best BLAST matches (or as many as alignability allows) is a good starting point for a more robust examination of the taxonomic affiliation of the query sequence (cf.
Anyone using the public sequence databases to pursue low-quality entries in a newly generated dataset will sooner or later find low-quality entries also in these databases. When skimming through BLAST hit lists, for instance, one regularly sees entries whose taxonomic annotation simply has to be wrong for one reason or the other - a single Betula (birch) in a list of Amanita (fly agaric), for instance. It is easy to feel that some mistakes are so far off and absurd as to be harmless. In reality they are harmless only to a limited number of people, namely those with a relevant taxonomic background; with a reasonable insight into how BLAST operates; and with enough time on their hands to interpret their sequence similarity searches manually. Everyone else may be in harm’s way. We did an informal evaluation of 20 fungal ITS sequences whose taxonomic annotation was off at the ordinal or class level by simply running the accession numbers through Google. Three of the sequences (15 %) had been used under their original (incorrect) name in at least one other scientific publication than the one through which they were released. Even taxonomic experts would be hard put to spot many such derived mistakes since they are published one level removed from the original data, suggesting a route through which errors and mistakes can be cited and re-cited enough to eventually be accepted as truths. There is thus every reason to take some form of action when one comes across a public DNA sequence associated with significant error.
The present document brings together a set of guidelines, recommendations, and observations towards identifying severely compromised sequences before they are put to scientific use. While they were written with the non-bioinformatician in mind and aim to be non-technical and straightforward to apply, we still believe they are powerful enough to have prevented the deposition of the vast majority of severely compromised fungal ITS sequences in the public sequence databases, had they been applied at the time of sequence accessioning. Importantly, however, these guidelines would not have caught all cases of badly damaged sequence data. Thus, the application of the principles presented here will not guarantee - but rather just increase the chance - that the dataset at hand will be of reasonable standard after processing. Furthermore we would like to stress that these guidelines offer little in way of fine-grained authenticity and reliability. Misidentification among closely related species, somewhat reduced levels of general sequence read quality, and base-inflation in homopolymer regions are all examples of problems that are only partly addressed by this document. We certainly do not want our guidelines to be used as replacements for more advanced, technical solutions; we rather hope that they will be used by those who, for one reason or the other, do not have access to or would not consider running any advanced, technical solutions in the first place (e.g., Appendix).
Our guidelines come with no other software requirement than a web browser. They still require something else of the user too: a critical, inquisitive, and perhaps imaginative mind. It would seem impossible to lay down firm rules to which all high-quality sequences would comply and that all low-quality sequences would violate. Rather the user should expect to find herself in situations where the user herself is the best arbiter of what is correct and what isn’t. Although such a situation would not be unfamiliar to anyone in systematics or taxonomy, we would still like to point out the importance of common sense in pursuing broken sequence data. The present authors spent considerable time trying to make this document as rich and multi-faceted as possible, but it goes without saying that additional, relevant observations and advice are to be found among the remaining members of the scientific community. We hope that anyone in the position to improve or add to the present set of guidelines will take the time and opportunity to do so. The potential outlets are many and range from the “Add comments” feature of the present journal to separate publications in this or any other journal. The ever-increasing weight assigned to molecular data in mycology - and the life sciences as a whole - suggests that any such move may have positive ramifications extending far beyond the datasets of each individual user.
RHN acknowledges financial support from Swedish Research Council of Environment, Agricultural Sciences, and Spatial Planning (FORMAS, 215-2011-498). LT and UK received financial support from ETF grants 8235, 9286, and FIBIR. EK acknowledges support from the Life Science Area of Advance at Chalmers University of Technology, the Swedish Research Council (VR), and FORMAS. CLS was supported in part by the Intramural Research Program of the NIH, National Library of Medicine. TMP acknowledges financial support from the Government of Canada through Genome Canada and the Ontario Genomics Institute through the Biomonitoring 2.0 project (OGI-050). AJ acknowledges financial support from Department of Energy’s Biological and Environmental Research Program (Award #ER65000). Yann Bertrand and Chris Quince are acknowledged for valuable input on the manuscript. Support from the Gothenburg Bioinformatics Network is gratefully acknowledged.
Technical considerations. (doi: 10.3897/mycokeys.4.3606.app) File format: PDF.
Explanation note: Discussion on sequence quality and reliability assessment for the more technically inclined user.